
The most interesting physical phenomena don’t occur in the bulk of a material, but right at the surface. Take two insulating oxide perovskites, for example: at their interface you encounter superconductivity, field-tunable metal–insulator transitions, and the formation of 2D electron gases, which may hold the key to the use of these materials in scaled-down electronic devices.
LaAlO3/SrTiO3 is a favored oxide pair, though its practical application is still far off due to the low electron mobility of the gases based on this couple. One proposed solution to this low mobility is δ-doping at the interface, which has been shown to improve the electron mobility of LaAlO3/SrTiO3-based devices from its poor starting point of 6–10 cm2 V−1 s−1.
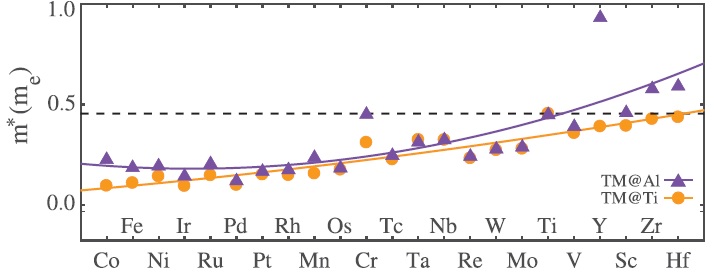
The dependence of effective mass on dopant identity for the oxide heterostructures.
To better understand how doping can improve the mobility of the 2D electron gas at such an interface, researchers at the University of California, San Diego, performed electronic structure calculations using density functional theory to explore the band structure, orbital populations, the electron effective mass, and the interfacial energy change for twenty-three different transition-metal dopants, spanning groups III to X on the periodic table.
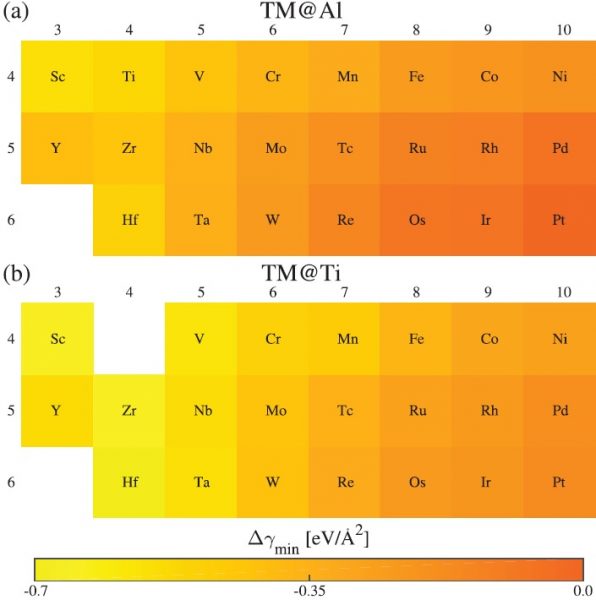
Dependence of minimum interfacial formation energy change on dopant identity for the different heterostructures.
They looked at the thermodynamic stability of each heterostructure. The energetic stability of the system relates to the range of electron mobilities that can be reached and says something about the experimental conditions required to achieve them. A negative value for the energy change associated with the interface suggests that the doped heterostructure would be favorable compared to the undoped heterostructure, which was found to be true of all but one system. A clear trend was observed, with more negative values calculated for group III dopants and then values getting closer to zero as they moved across the periodic table toward group X.
“We find that there exists a trade-off between achieving light effective mass bands and obtaining energetically favorable structures for the δ-doped LaAlO3/SrTiO3 heterostructures,” the researchers indicate in their Advanced Materials Interfaces paper. See the details of their findings here.
Interested in perovskites or electron mobilities? How about these recent articles:









