
The temporal evolution of a biological system is the result of complex interactions, which may involve a wide variety of different biochemical components such as proteins or metabolites. In recent years, these systems have often been described through mathematical models that translate these interactions into equations or rules. Once a mathematical model is defined, a computer simulation is the realization of the temporal evolution of the system by using ad hoc simulation algorithms.
Nowadays, the mathematical models and their computer simulations are essential tools in investigating biological phenomena as they provide a fast and convenient framework to systematically analyze the system, to explore its dynamics, and to develop and test new hypotheses. Moreover, these model simulations offer new insights into existing experiments, support the design of new studies, and, ultimately, assess the suitability of specific molecules as novel drugs or therapeutic targets.
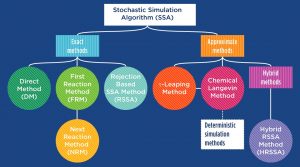 Simulation algorithms are often classified as stochastic or deterministic, depending on the level of approximation they introduce into the simulation of the system dynamics. Stochastic algorithms are the most accurate and they are able to reproduce the random and discrete nature of the biological system. However, they can become prohibitively slow when applied to large biological systems, where thousands of reaction firings can happen per time unit.
Simulation algorithms are often classified as stochastic or deterministic, depending on the level of approximation they introduce into the simulation of the system dynamics. Stochastic algorithms are the most accurate and they are able to reproduce the random and discrete nature of the biological system. However, they can become prohibitively slow when applied to large biological systems, where thousands of reaction firings can happen per time unit.
To circumvent this problem, approximate methods have been introduced to decrease the run-time at the cost of sacrificing accuracy. Among the approximations, the deterministic approaches completely neglect the random noise of these biological processes by providing an average behavior.
The authors of a WIREs Systems Biology and Medicine review present an extended overview of the main stochastic exact and approximate algorithms. They introduce the problems and the reasons behind the need for different simulation strategies and they guide the reader through the pros and the cons of each method with respect to the others. The aim of the review is not only to compare and describe the algorithms, but also to provide a practical guide to their implementation by including the pseudo codes.
In addition, the authors include the test case of the sphingolipid metabolism, a family of bioactive lipids involved in several regulation processes, as well as in the development of different diseases. The use of three simulation algorithms with different levels of accuracy in this example exemplifies how different strategies may unveil different insights and support different biological findings.








