
The material universe is all about bonding! Everything in our natural world is made from molecules and these molecules are held intact by chemical bonds. Bonding is fundamental not only for the basics of chemistry and biology, but for everything we experience in our daily lives; the molecules in our food, the proteins in our body, the gas in our car, the oxygen we breathe all exist in their current forms because of chemical bonds. Scientists will of course be familiar with two fundamental bonding types: covalent and ionic bonding interactions, but a new type of bond called charge-shift bonds (CSBs) has long been overlooked.
A brief history of bonding
At the time when bonding theories were being formulated in the 20th century, the electron-pair bond was, and perhaps still is, considered the major form of bonding in molecules. It was formulated more than 100 years ago by Gilbert Newton Lewis, as a pair of electrons which holds two atoms. This idea was further articulated by chemists who found it useful to explain the three-dimensional shape of molecules as a result of the repulsion between the electron-pairs that surround a given atom (figure below). This formulation has allowed us to imagine new molecules by taking atoms and “clicking” their electrons into pairs to form bonds. One could therefore use this seminal idea and draw on a piece of paper an entire chemical universe.
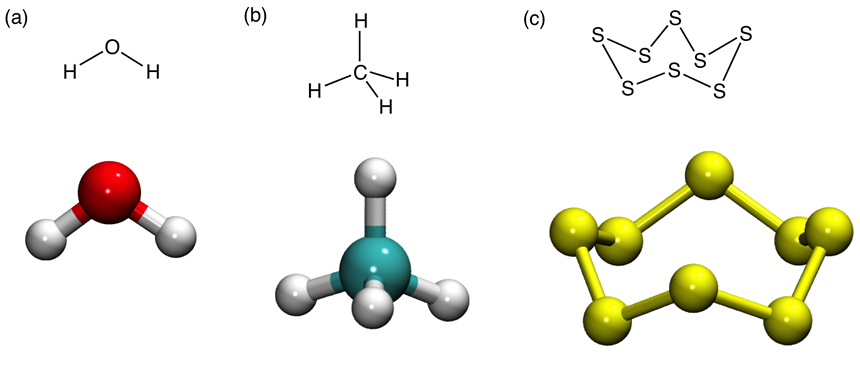
Lewis was originally driven to explain the different observed behaviors of compounds that were made in two communities of chemists. In simplified terms, some molecular types (made of the atoms, C, H, N, O) did not conduct electricity, while other types (made of metallic atoms and e.g., O, Cl, F) conducted electricity e.g., when in solution in water.
Lewis constructed his bond such that the electron pair existed anywhere between the two atoms, sharing it, and envisioned a scale of the degree of this shared bonding that stretches between a shared pair — which was later called “covalent” as in the C-H bond of CH4 — and an atom-possessed pair — later called “ionic” as in Na+ Cl–.
In the early 1930s, another great chemist named Linus Pauling went to Europe to learn about a new, emerging theory of matter called quantum mechanics, and was a witness to the first theoretical description of a covalent bond in the H2 molecule. The idea was put forth by the young postdoctoral fellows of Erwin Schrödinger, Walter Heitler and Fritz London, who invented the new theory of bonding called valence bond theory. Being a chemist, Pauling was very excited because he immediately recognized that Heitler and London’s theory was a mathematical formulation of the covalent bond. He and his contemporary scientist, John Slater, extended valence bond theory, and in addition to the covalent form postulated by Heitler and London, for an A-B bond they added the two ionic forms: A+ B– and A– B+, which are allowed to mix into the covalent form.
To simplify matters, Pauling assumed that the homonuclear or homopolar bonds (bonds between identical atoms such as H2 or F2) are all 100% covalent, whereas polar bonds, which are bonds between two different atoms that have, in simplified terms, different “electron attracting” abilities (or electronegativities), are a resonating mixture of covalent and ionic components. As the mixing of these two components — in which the electron pair fluctuates between the covalent and ionic forms — is associated with a so-called “resonance energy”, this interference of two pieces of the wave function is always stabilizing. As such, Pauling’s simple model readily explained why polar bonds are, as a rule, stronger than homopolar ones in which the resonance energy was assumed to be nil.
Despite great progress made over the past several decades in the development of advanced computational tools of quantum chemistry, these seminal ideas put forth by Pauling, London, and Heitler have survived in mainstream chemistry and are still taught in classrooms around the world. Yet, over time, some unexplained phenomena have been gradually building that could not be explained by Pauling’s intuitive model as it is oversimplified and possibly incomplete.
At some point, we felt that a rigorous quantum mechanical evaluation of Pauling’s assumptions was needed.
Enter charge-shift bonds
Our journey began in 1984, when the two of us were working in a laboratory studying theoretical chemistry in Orsay, where one of us (P.C.H.), was a researcher in the laboratory, and the other (S.S.) came for a sabbatical year. Being both fans of valence bond theory, we got together and devised a computer protocol that would enable us to run reasonably accurate valence bond calculations of molecules. It was excruciatingly slow and difficult but it allowed us to calculate the first set of simple bonds using valence bond theory and publish our work in 1991.
One of the molecules we modeled was F2 (di-fluorine gas), which has an electron pair that holds the two F atoms together. The molecule showed a strange behavior: in the eyes of every chemist, F2 was by definition a covalent bond since the two bonded atoms are identical to each other. However, when we used our valence bond protocol and calculated the molecule, we found that its purely covalent form — in which each F atom equally contributes one electron to the bond — was repulsive at all F—F distances! Meaning, the covalent form gave no bonding whatsoever, indicating that a covalent bond should not exist. The ionic forms, on the other hand, seemed unfavorable because of their very high energy. Nevertheless, when they were included in the calculation, lo and behold, the molecule became bonded by a significant amount (38 kcal/mol), in agreement with experimental data.
These results were exciting and intriguing and turned what was thought to be a conventional bond on its head. We then realized that the factor that was leading to a bonded electron pair in this molecule was in fact a quantum mechanical phenomenon in which an interference between the two components of the wave function is responsible for the entirety of bonding; in the valence bond lingua, this is the resonance energy between the covalent and ionic terms. This is symbolically represented by the double headed arrow between the forms (covalent on the left and ionic on the right):
F•—•F ⟺ (F+ F– + F–F+)
In quantum mechanics, resonance between different forms always lowers the energy, and is the root cause for the bonding in F2.
This was precisely what was missing in the oversimplified approximation made by Pauling who assumed that this covalent-ionic resonance energy is zero in all homonuclear bonds. Thus, if we use the common energy units in chemistry (kcal/mol), then the resonance value for the H-H bond is less than 10 kcal/mol and for the ionic bond Na+Cl– it is 5.4 kcal/mol. In comparison with these values, for F-F the resonance energy is an order of magnitude larger at almost 70 kcal/mol. But more so, this large quantity overcomes the repulsion between the F• atoms in the covalent form and endows the molecule with substantial bond energy and strength.
A breakthrough and a new type of bond
Seeing more and more cases similar to F-F, we began to realize that we had discovered, quite accidentally, a new form of electron-pair bond and called it the charge-shift bond family on the advice of our friend, the late Edgar Heilbronner. Thus, charge-shift bonds are defined as those bonds which exist primarily due to the resonance energy between the covalent and ionic forms. This situation is at variance with covalent bonds or ionic bonds, where bonding mainly arises, respectively, from the covalent and ionic wave functions.
Our earlier computational investigations showed that charge-shift bonds have a variety of properties, which distinguish them from their covalent and ionic congeners, and this immediately explained why some bonds did not fit in Pauling’s model and were formerly viewed as intriguing exceptions.
Charge-shift resonance shapes molecular properties
It is well accepted and experimentally rooted that, as a rule, classical covalent bonds (e.g., H-H, C-C and so on) display an accumulation of electron density in between the atoms. However, some seemingly covalent bonds depart from this rule, and display zero-accumulation or even a depletion of density, as in the case of difluorine (F-F). More generally, the family of such bonds, sometimes called “no-density bonds”, coincide with the charge-shift bond family as defined by valence bond theory.
The probability of presence of an electron pair can also be visualized by the method of “electron localization functions” (ELF), as shown in the following figure. For the C-C bond of ethane (H3C-CH3), on the left-hand side, one clearly sees an accumulation of density in green in the middle of the C-C bond, while the lilac volumes figure the C-H bonds. For F2, on the left-hand side, there is a dismal density in green in the middle of the F-F bond, while the orange volumes figure the lone pairs. Thus, H3C-CH3 is a classical covalent bond whereas F-F is a typical no-density bond.
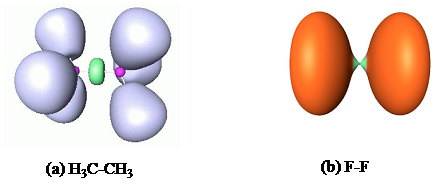
Another example concerns the chemical reactivity patterns of covalent bonds vs. charge-shift bonds. The carbon-chlorine bond (e.g., in (CH3)3C-Cl) easily undergoes dissociation to two ions in water, leading to the very common carbenium ion ((CH3)3C+) and to the associated ionic chemistry. This is because the C-Cl bond is quite polar and that its ionic C+Cl– component is strongly stabilized by solvation. Even easier is the dissociation of the sodium-chlorine bond, leading to solvated Na+ and Cl– ions, which constitute salty water. By contrast, silicenium cations (e.g., (CH3)3Si+) are extremely rare in condensed phases, despite the fact that the polarity of the Si-Cl (e.g., in (CH3)3Si-Cl) bond is in-between those of the C-Cl and Na-Cl bonds.
More generally, the ionic chemistry of Si-X compounds (X = very electronegative group) is extremely rare. This puzzling particularity of the Si-X bonds becomes clear when one realizes that these bonds are charge-shift bonds, whereas C-X ones are not. Thus, the charge-shift resonance energy of (CH3)3Si-Cl amounts to 51 kcal/mol. More so, this quantity is preserved in solution and stabilizes the (CH3)3Si+Cl– ion-pair, thus fully entering the activation barrier for bond heterolysis (recall that this stabilization is lost as the two ions are pulled apart to infinity). It follows that the silicenium ion is very “sticky” and its free form is rare in solution or in the solid state.
Another manifestation of the charge-shift resonance energy can be extracted from experimental measurements of the energy required to activate a molecule and break its bond. Based on the figure below, imagine that atom X•, which has an unpaired electron (like F• or H•), impinges on a molecule that has a bond H-X’ (X and X’ are identical and the primed X serves simply as a flag to distinguish it from the right-hand side X). As X• collides with H-X’ from the H side, the energy increases because the H-X’ bond gets gradually stretched. At some point, shown in example (a) below, when the distances of the two X atoms from the central H are identical, the energy rise reaches its maximum. This X•••H•••X’ molecular entity is called a transition structure, and as the H•••X’ bond further elongates, while X•••H is shortening, the energy starts decreasing until the two X atoms are exchanged, giving rise to X-H, in which X and H are bonded by an electron-pair bond, and the free atom X’•. The energy required to reach the transition structure is called an activation barrier. In a similar fashion H• can impinge on the X-H’ molecule from the X side, thus forming the transition structure H•••X•••H’ (b) that exchanges H by H’ to form H-X and H’•.
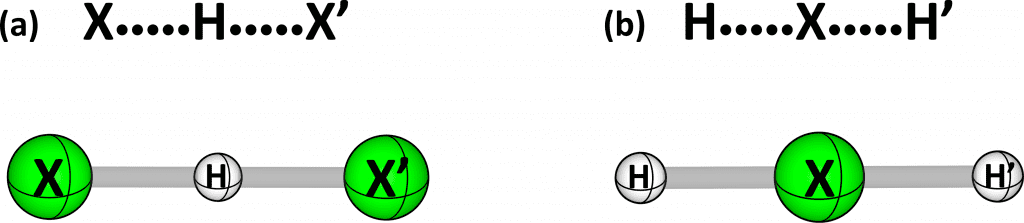
Although in both reactions one breaks the same H-X bond, these two reactions have very different activation barriers. As we showed in 2006, the barrier for the reaction in the (b) is much higher than that of the reaction in (a). This relation of the barrier arises due to the greater loss of the charge-shift resonance energy in the transition structure for reaction (a) compared with (b). When X is F, the H-F bond is a charge-shift bond, having a very large charge-shift resonance energy (88 kcal/mol), and hence, the difference between the barriers of the two reactions is very large, much larger than that when X is Cl, Br or I, which form polar-covalent bonds with much smaller charge-shift resonance energies.
Moreover, the difference between these barriers is given as ca. 25% of the charge-shift resonance energy of the H-X bond! This means that the charge-shift resonance energy, which is a brainchild of valence bond theory, is in principle a measurable and a computable quantity, which can be accessed independently of the theory one uses.
Yet another phenomenon that does not fit into Lewis-Pauling’s model is the existence of bonds involving noble gas atoms, for example Xenon in XeFn (n=2,4,6), whereas other noble gas atoms (e.g. He, Ne) have a small propensity for bonding. Besides, other hypervalent compounds such as PCl5, SFn (n = 4,6), and so on, are also stable, whereas many other ones are unstable. As a general rule, we could show that the only atoms that can form hypervalent molecules are those that can form charge-shift bonds with other atoms.
An ever-changing landscape
After carrying out many more valence bond theoretical studies — summarized in our recent essay — we have been able to establish that the territory of CSB involves homopolar bonds of compact electronegative and/or lone-pair-rich elements, heteropolar bonds of these elements among themselves and with other atoms, no-density bonds, three-electron bonds (as exist in, e.g., the dioxygen that we breathe), dative bonds and hypercoordinated molecules. A few weeks ago, we showed that charge-shift bonding is also common in transition-metal elements, which have 3d orbital-shells, and the trends in these bonds, can be easily understood and predicted.
Chemistry is constantly changing and it’s very exciting to challenge something so fundamental to the field. As a result of this “missed family” of electron-pair bonds, we can finally rationalize unexplained features and phenomena, thus, enriching our understanding of molecular structures and reactivity.
Written by:
Sason Shaik is a Saerree K. and Louis P. Fiedler Emeritus Professor of Chemistry at the Hebrew University. His main interests are in bonding, chemical reactivity, metalloenzymes, and electric field effects in chemistry. He uses valence bond theory as a conceptual frame, and has developed a number of new paradigms and concepts using this theory. He is a Schrödinger Medalist (2007); an August-Wilhelm-von-Hofmann-Medalist (2012); a member of the International Academy of Quantum Molecular Science (2015); a Gold Medalist of the Israel Chemical Society (2017). He writes essays and poetry.
Philippe Hiberty is an Emeritus Director of Research at the Paris-Saclay University. In 1994, he elaborated the “breathing orbital valence bond method“, devised to combine compactness and accuracy for valence bond wave functions. He now applies it to challenging problems in photochemistry. His research interests are, among others, in the application of quantum chemistry and valence bond theory to fundamental concepts of organic chemistry.
______
This highlight is based on our latest essay published with our colleagues, Benoit Braida, David Danovich, John M. Galbraith and Wei Wu. Reference: S. Shaik, P. Hiberty, et al. ‘Charge‐Shift Bonding: A New and Unique Form of Bonding.’ Angew. Chem. Int. Ed. (2019). DOI: 10.1002/ange.201910085









