Absorption, distribution, metabolism, and excretion (ADME) studies are essential to the drug discovery process. These studies provide the pharmacokinetic and pharmacodynamic properties of potential drug candidates and offer crucial information on prospective safety and efficacy.
Among these studies is the mouse-liver microsomal (MLM) assay, a key pharmacological assay able to predict metabolic stability and a required aspect of any investigational new drug (IND) application. The ability to predict the results of ADME assays, including MLM, using computational tools/models would expedite the drug discovery process immensely while also decreasing costs.
In a recent review published in WIRES Computational Molecular Science, Dr. Yufeng Tseng and his team from National Taiwan University compare published machine learning models predicting MLM stability to a model constructed using deep learning methods.
Deep learning models, which fall into a subset of machine learning methods, make use of artificial neural networks originally inspired by biological neural networks. Neural networks are comprised of artificial neurons, or nodes, connected to a web of other nodes through “edges,” allowing these artificial neurons to receive and transmit information to each other.
Typically, nodes are organized through distinct layers: input nodes that transmit information through multiple hidden layers that help the model to learn which features are associated with which outcomes, and an output layer that gives the final classification of a data piece. The process of learning for neural networks involves changing the weights and biases of certain nodes such that certain features detected within the input layer are correctly classified within the output layer every time. The benefits of deep learning models over traditional machine learning models are vast, including quicker training times, more efficient use of computation, and higher predictive accuracy on a wider variety of datasets.
The recent development of graph convolutional neural networks (GCNs) has furthered the practicality of deep learning models in the realm of computational chemistry. Due to the inherent graphical nature of molecules (with atoms representing vertices of a graph and bonds representing edges), graph convolutional methods can directly input graphical features of molecules into the neural network.
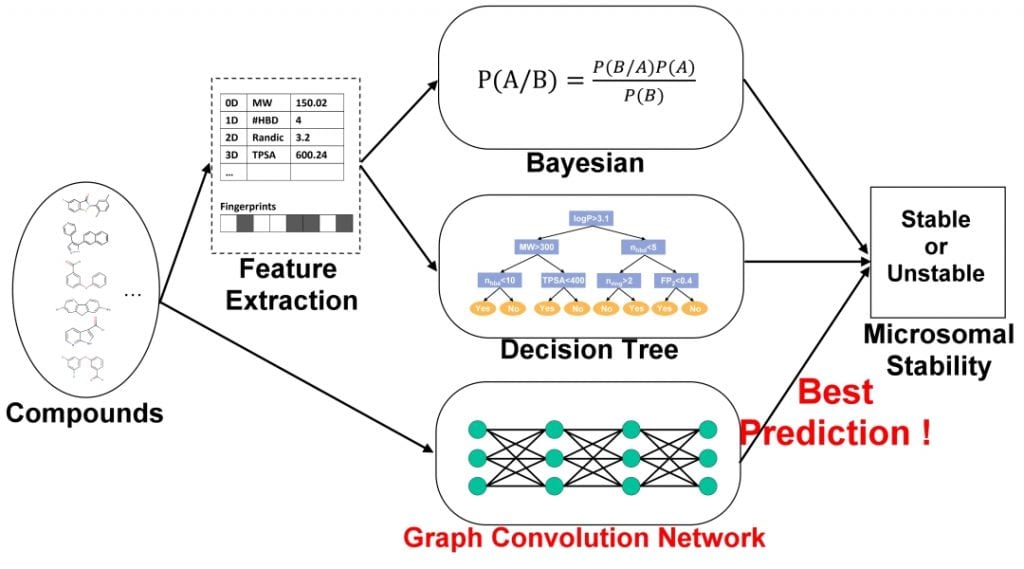
In contrast to the traditional representation of molecules that utilize off-the-shelf fingerprint software to compute fixed-dimensional feature vectors as inputs, graph convolutional methods are able to develop their own fingerprints that are flexible and machine optimized. This has been shown to provide stronger predictive performance, increased interpretability of similar features, and more streamlined feature representation compared to fixed-fingerprint features, reducing computational efforts and regularization requirements.
The researchers showed similar results in a GCN developed for the purpose of MLM stability prediction. Their GCN, trained on a dataset identical to a state-of-the-art Bayesian classifier, provided stronger predictive performances against both validation and test sets when compared to said Bayesian. The increased prediction was shown in its higher accuracy, sensitivity, specificity, and AUC ROC scores, with the overall accuracy increasing 6.6% in the test set (83.3% vs 76.7%) and 22.6% in the validation set (78.8% vs 56.2%). The ROC scores saw similar results as the GCN showed +0.076 (0.861 vs 0.785) in the validation set, and +0.160 (0.864 vs 0.704) in the test set.
They also developed a decision tree (C5.0 classification model), constructed through a total of 1875 PaDEL-descriptors and 12 types of fingerprints and trained on the same identical dataset. When model performance was compared against the GCN, the C5.0 saw a level of overfitting in its prediction of the validation set molecules and its accuracy drastically diminished. Previously published machine learning models, including more Bayesian classifiers and random forest models, also showed decreased accuracy against the GCN.
In both the Bayesian and C5.0 decision tree, fixed-descriptors developed through alternative programs were used as inputs into for the model. While many factors may have contributed to the decreased predictive performance compared to the GCN, it is a major limiting factor that machine learning models must include an initial step of manual feature extraction. Graph convolutional methods bypass this, creating more efficient and precise features of molecules through simply using molecules themselves as inputs into their network.
With improvements to the theory surrounding graph convolutions and the development of more efficient algorithms, GCNs may come to be a pivotal tool in drug discovery pipelines, expediting the process and vastly streamlining efforts.
Article written by Alex Renn, Bo‐Han Su, Hsin Liu, Joseph Sun, and Yufeng J. Tseng
Reference: Alex Renn et al. ‘Advances in the prediction of mouse liver microsomal studies: From machine learning to deep learning,’ WIREs Computational Molecular Science (2020). DOI: 10.1002/wcms.1479