
Macromolecules—giant natural or synthetic molecules—with intricate compositional, architectural, conformational, and configurational features, play a vital role in all aspects of life. Accordingly, from both scientific and industrial standpoints, manipulating and fine-tuning the microstructure of macromolecules to tailor their final properties is of paramount importance.
Modifying and adapting the microstructure of macromolecules by the most appropriate polymerization recipe has been regarded as a challenging effort. “Cracking” the complexities of the interrelationships between a polymerization recipe and the microstructure of the resulting macromolecules is a very complicated, multi-objective optimization problem that has been attracting increasing attention over the last decade.
Despite the fact that molecular simulation techniques like Kinetic Monte Carlo have been successfully applied to gain detailed micromolecular-level information (about the microstructure of macromolecules), they are mostly expert in obtaining microstructural signatures of the macromolecules in response to process input conditions. In many applications, however, the desired properties of the macromolecules can be specified in advance, and one should ideally be able to move backwards and define the reaction conditions required to obtain the corresponding microstructures. In other words, dialling in a certain set of desirable product properties and subsequently obtaining feasible suggestions for a set of input factors to engender such properties (via some modelling/simulation software packages) is of great practical and economical significance.
To synthesize the predesigned microstructures, i.e., reverse simulation moving from the macromolecular microstructure (as the problem input) to the reaction conditions (as the problem output), requires searching a large multivariable space for the optimum conditions to synthesize the predesigned microstructures.
Such a tedious task for traditional modelling/simulators can be done by providing typical molecular simulation approaches with the capability to make decisions. Thus, amalgamating Kinetic Monte Carlo and Artificial Intelligence-based modelling/optimization techniques, a novel intelligent simulator—referred to as Intelligent Monte Carlo (IMC)—has been proposed by an international collaboration between Alexander Penlidis (University of Waterloo, Canada), Krzysztof Matyjaszewski (Carnegie Mellon University, Pittsburgh, USA), and several other leading experts.
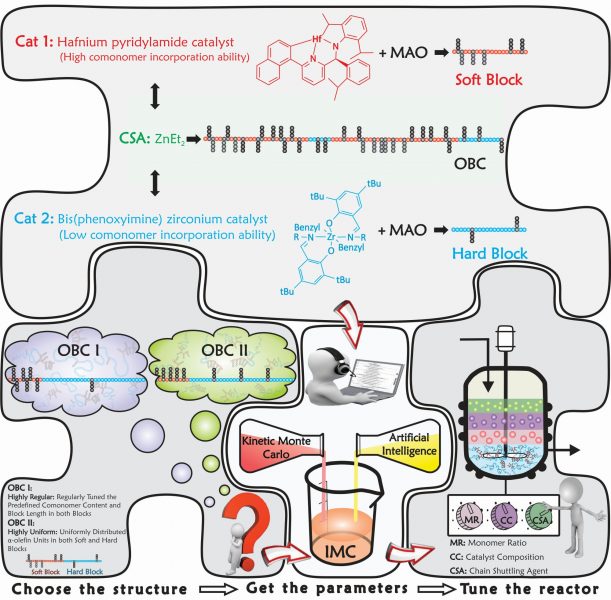 IMC is a versatile and powerful intelligent tool, which has been successfully put into practice to effectively address the product quality control issues mentioned above. In fact, the newly developed IMC technique can thoroughly handle complex macromolecules. It is like drawing or imagining any intricate hypothetical macromolecule on paper first, then bringing the virtually drawn or designed macromolecule into existence via the intelligent IMC simulator.
IMC is a versatile and powerful intelligent tool, which has been successfully put into practice to effectively address the product quality control issues mentioned above. In fact, the newly developed IMC technique can thoroughly handle complex macromolecules. It is like drawing or imagining any intricate hypothetical macromolecule on paper first, then bringing the virtually drawn or designed macromolecule into existence via the intelligent IMC simulator.
IMC starts from the simulation of complex polymerizations; learns and identifies the dependency of microstructural features on the operating/recipe conditions; finds the closest set of operating conditions that lead to control and optimization of several microstructural factors simultaneously; and checks for the authenticity of the predictions by synthesizing or “designing” the target macromolecule.
This approach provides a new platform for identifying complex reaction conditions to produce and tailor-make materials with precisely predefined microstructures. In fact, IMC facilitates the development of meaningful structure–property relationships.









