The way molecules interact with one another in solutions is only partially determined by direct attractions or repulsions between them. In addition, the overall binding affinity (the tendency of molecules to form complexes or aggregates) of solvated molecules can be strongly influenced by interactions with the solvent.
In aqueous solutions, the most well-known manifestation of such solvent-mediated interactions are hydrophobic forces, which describe an attractive mean force between nonpolar molecules in water due to their tendency to minimize their exposure to the solvent. Additionally, solvent-mediated interactions, in particularly hydrogen bond bridges, also play a key role in surface interactions.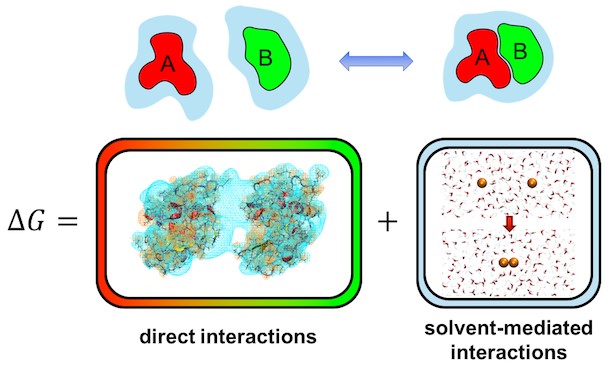
The key quantity that provides a quantitative description of solvent-mediated interactions is the solvation free energy. A review in WIREs Computational Molecular Science by Matthias Heyden describes techniques that quantify the solvation free energy and its enthalpic and entropic contributions from all-atom molecular dynamics simulations in an explicit solvent. Especially the ability of these methods to spatially decompose solvation free energies into local contributions allows us to understand the solvation of complex molecular surfaces in microscopic detail. The latter is required for accurate predictions of solvent-mediated intermolecular forces that result from changes in the solvation free energy when molecules approach each other in solution.
The methods described in this Advanced Review can provide vital information that improve our ability to predict the affinity of drug candidates to their biomolecular targets. In addition, the microscopic information on solute-solvent interactions obtained from this analysis may play a crucial role for the development of improved implicit solvation models that allow for computationally efficient simulations of interacting molecules in solution.
Kindly contributed by the Author.