
Polymer melts, though studied extensively by theory, computer simulations, and experiments still pose many questions. While equilibrium properties are reasonably well understood, i.e. conformations of chains or basic rheological parameters, many non-equilibrium issues, such as conformational properties during processing (different strain rates, shear geometries etc) lack a microscopic understanding. Such an understanding in turn can lead to important insight and hints towards optimized processes or experiments. Computer simulations, employing advanced hardware as well as highly efficient software are becoming powerful enough to tackle such problems for highly entangled polymer melts. Though so far mostly employed for simplified models, modern scale bridging approaches make this possible also for models containing chemical details. One of the advantages of such simulations is the fact, that on has perfect control over the studied system, e.g. the polydispersity.
 Though opening exciting options, there is still the problem of reference system or system from which the simulations start. Typically, for a full equilibration of a polymer melt within a computer simulation, simulations up to the polymer relaxation time might be required. Among others this is a consequence of the non-crossability of the polymers, which at the same time is responsible for the interesting and rich physics of such melts. This is a major drawback, since this time typically is of the order of the simulation time of the whole project. Thus there have been ample activities to circumvent the slow physical path of equilibration.
Though opening exciting options, there is still the problem of reference system or system from which the simulations start. Typically, for a full equilibration of a polymer melt within a computer simulation, simulations up to the polymer relaxation time might be required. Among others this is a consequence of the non-crossability of the polymers, which at the same time is responsible for the interesting and rich physics of such melts. This is a major drawback, since this time typically is of the order of the simulation time of the whole project. Thus there have been ample activities to circumvent the slow physical path of equilibration.
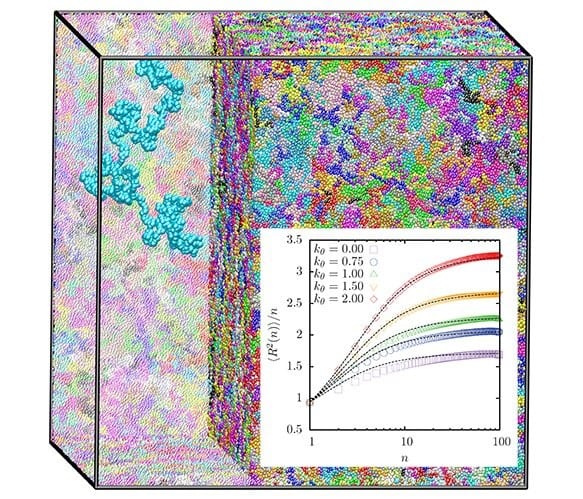
Researchers from the Max Planck Institute for Polymer Research now present an improved methodology, which only requires local equilibration and which very efficiently leads to equilibrated melts of long polymer chains. The longest chains presented as test cases in this study contain up to 45 entanglement lengths. Mapped onto PS, that would correspond to polymers of a molecular mass of about 800,000. To demonstrate the prospects of the approach, the equilibrated ensembles are used to examine the entangled state of the simulated polymer melts. They demonstrate that the approach efficiently leads to equilibrated ensembles of 1000 chains of up to 2000 beads of bead-spring polymer melts. The figure illustrates such a system and shows characteristic intra-chain distances, which are very sensitive measures of equilibration. A master curve for the estimating entanglement length for polymer melts with different chain lengths and persistence lengths is provided and can serve as a reference for other investigations.










