
Materials science is concerned with engineering and characterizing new materials for any sort of applications: from transportation to computing to medicine, to name a few. It’s method of inquiry is often guided by computational models; however, the bulk of the investigations are done experimentally.
The future will witness a gradual shift in which computational models will play a progressively larger role in identifying new materials for specific purposes. Eventually, we will transition to a second-generation method of inquiry, a.k.a “Lab 2.0”, a type of materials research and engineering that carries out most scientific investigations with computational models, employing experiments only for the last few steps before commercialization i.e., characterization, verification, and synthesis of a shortlist of materials candidates.
While researchers are still a ways away from fully realizing Lab 2.0, several advances are moving us towards it: 1) computer speed increases every year; 2) accurate computational methods are becoming ever-more sophisticated for complex systems, such as those based on quantum mechanics like Density Functional Theory (DFT)), and 3) there is a growing community of scientific software developers supporting scientific software innovation.
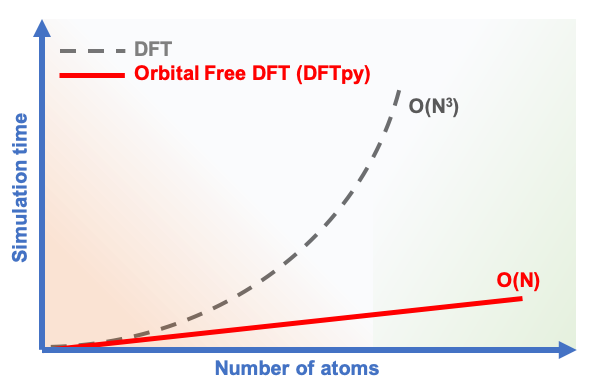
Among the available computational materials science tools based on quantum mechanics, Orbital-free DFT is emerging as both computationally cheap and accurate in modelling real-world scenarios. As shown by several recent studies, this technique is promising because it can model systems of realistic sizes, such as entire electronic devices. An impressive capability from a first-principles perspective in which the model has no specific knowledge of the system, and it is only based on the basic laws of physics, as well as quantum-mechanical and atomistic models!
A Rutgers University team led by Professor Michele Pavanello developed an orbital-free DFT software, named DFTpy, which achieves record efficiency and is able to approach systems composed of millions of atoms with a single processor. “This is a record for any DFT code”, says Pavanello. Even though Lab 2.0 may still be far away, advances in orbital-free DFT are making important steps towards it.
DFTpy is written entirely in Python 3, an emerging programming language widely used for web development, machine learning, databases, and recently also scientific computing. Python provides developers with a low barrier to entry, a crucial aspect for a software developed in academia. “Thanks to its Python codebase, DFTpy offers a great framework where we can easily develop algorithms for modeling how materials respond to external stimuli and run comprehensive and atomistic simulations based on orbital-free DFT” says Kaili Jiang, one of the study’s co-authors.
The second generation of DFTpy is already being developed. “Typical DFT algorithms are too slow for modeling nanoscale systems,” says Xuecheng Shao, the lead author of the study (see figure). He is now developing “embedded DFTpy” (eDFTpy) in collaboration with the Andreussi group at the University of North Texas. eDFTpy will leverage the successes of DFTpy to implement a suite of software for device-level simulations, which typically involve 10 million to 1 billion atoms. Shao goes further: “The combination of orbital-free DFT and other techniques like embedding is what is really needed to model complex systems.”
Orbital-free DFT realized in the DFTpy framework “will allow us to predict mechanical and photophysical properties of device-level systems from first principles – a much needed state-of-the-art for materials science and engineering, getting us even closer to Lab 2.0.,” says Pavanello. So, stay tuned!
Reference: Xuecheng Shao, et al. ‘DFTpy: An efficient and object-oriented platform for orbital-free DFT simulations.’ WIREs Computational Molecular Science (2020). DOI: 10.1002/wcms.1482









