Image credit: National Cancer Institute
Timing plays a crucial role in biological processes, and computer simulations have helped advance our understanding. Modelling pathways such as DNA synthesis and repair or trafficking cargo between different cellular compartments is significant in health and disease. They all require an intricate understanding at the molecular level, and as opposed to laboratory experiments, computer simulations can model and provide more detailed predictions for these processes cheaply and rapidly.
Molecular dynamics is a commonly used computational approach in which the physical movements of molecules are followed to better understand their structure as well as interactions taking place between them—such as during a chemical reaction. This allows researchers to explore the “conformational energy landscape” of a system over a fixed period of time, meaning they can watch the natural and dynamic evolution of a system in order to better understand it.
To obtain accurate predictions, molecular dynamics uses a stepping scheme, in which the system is approximated not in one whole step, but over thousands of smaller “timesteps”. This requires that each step must be small, and our current capabilities allow for steps that are about a femtosecond (10-15s) long.
However, many biomolecular events occur in the range of milliseconds and seconds. The time scale difference between the integration time step and the time of biological events implies that about 1012 integration steps must be completed to reach a millisecond-scale event.
Models of biological systems are large and frequently include hundreds of thousands of particles. Therefore, even the calculation of a single integration step is expensive. On advanced super computers, computation speeds are measured in microsecond biological time (10-6s) per day of calculation. A calculation of a single millisecond trajectory requires 103 days, or about three years. The need to obtain meaningful statistics (say, one hundred events of cargo trafficking) further increases computational complexity.
To overcome the challenges of these biophysical time barriers, a new theory and algorithm is required. In a recent paper published in the journal WIREs Computational Molecular Science, “Milestoning” was described for this purpose.
To appreciate the difference between conventional molecular dynamics simulations and Milestoning, consider an examination of a 100-mile-long road between city A and city B. One solution is to send a walker from A to B. The walker progresses about one mile an hour, and therefore the whole examination will take 100 hours.
This process can be sped up if 100 walkers are assigned to the same task. For example, each walker starts at a different position along the road, called a “milestone.” The 100 walkers travel only up to the next milestone, significantly reducing the amount of time required to cover the entire road: one hour versus 100.
An estimate for the overall time of one walker journey can be obtained by summing the times of the 100 walkers. The more walkers and milestones used, the more time-efficient the journey or the examination becomes.
In a biological/chemical context, the road from the previous analogy represents a reaction coordinate, where molecules move along a pathway during a reaction; from starting materials to products. While the road is a perfect-case scenario, molecules are not reliable walkers as they can move backward and forwards along the road (reaction coordinate) with probabilities to be determined.
The probabilities that molecules move to the next or previous milestones are estimated by sampling. We initiate several “walkers” at each milestone and record who went in each direction. For example, the number of molecules that went forward divided by the total number of “walkers” from a milestone is an estimate of the forward probability. The trajectories between the milestones are less than a nanosecond in length (10-9 s) and provide an efficient parallel algorithm to simulate the entire event. The algorithm running-time scales linearly with the number of milestones.
An additional complication is a synchronization at the milestones to ensure that the multiple short trajectories are equivalent to a single walker. This Milestoning theory patches the short trajectories at each milestone together by matching the incoming and outgoing fluxes of walkers. This means that the number of walkers leaving the milestone must be equal to the number of walkers arriving at it.
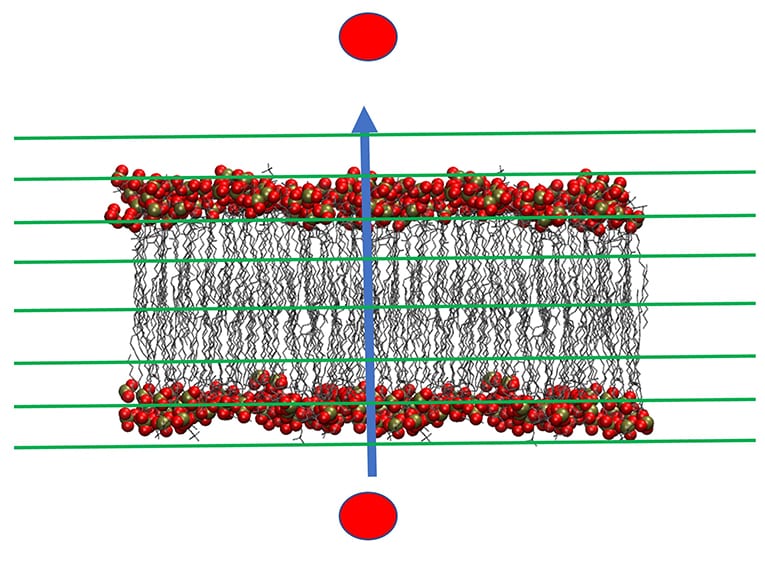
This strategy made it possible to accurately simulate many long-time biophysical processes that are not accessible through conventional molecular dynamics due to computational complexity. For example, with Milestoning we are able to simulate peptide permeation across membranes which biologically takes hours. A group of peptides, called cell penetrating peptides (CPP) play a significant role in translocation or the movement of a molecule across a bilayer. They pass efficiently and selectively through membranes of different cells or compartments and act as antibacterial and anticancer agents. Therefore, the molecular mechanism of their permeation is of considerable biological and medical interest. Milestoning now makes it possible to investigate these biological events at atomistic resolution.
A challenge for future studies is the rigorous determination of reaction coordinates as sometimes there is more than one way that connect the two cities. While still in its early stages, this approach could change the way in which we think about and understand biological systems.
Written by: Ron Elber, Department of Chemistry and the Oden Institute for Computational Engineering and Science, University of Texas at Austin
Reference: Modeling Molecular Kinetics with Milestoning, WIREs Computational Molecular Science (2020). DOI: