Given its ubiquitous roles, one would expect that a biological cell’s membrane should be well understood. After all, a membrane is what protects and regulates the cell’s interaction with its environment. It is the first point of contact through which material can be exchanged between the cell interior and its surroundings. While we do know a great deal about the structure and compositions of biological membranes, the actual mechanisms by which matter is transported in and out of cells across membranes is, surprisingly, not well established. Understanding how molecules can penetrate cells—naturally for better or worse, or by design for drug delivery—constitutes knowledge as priceless as human health.
A membrane consists of a thin lipid bilayer, which constitutes an hydrophobic barrier separating hydrophilic interior and exterior environments. This arrangement restricts the diffusion of polar molecules across the membrane. A textbook explanation of how material is exchanged through a membrane relies on the existence of pores and channels. While their formation has been inferred from a number of experimental and theoretical works, direct observation of such pores has been elusive. Some recent experiments have provided new evidence for permeation of membranes that is not compatible with the notion of pore formation—throwing up questions as to whether we understand this issue fully.
Clarifying molecular processes such as membrane permeation requires tools that can probe temporal and spatial scales beyond what can easily be achieved with current experimental techniques. This is where our computational methods come in handy. By using molecular dynamical and quantum mechanical computations, our group landed upon a fundamental new understanding of membrane dynamics with transformative potential for applications such as drug delivery. Our study, published in Advanced Theory and Simulations, started looking at a positively charged peptide traversing a cell membrane. The charged peptide experiences high barriers within the hydrophobic interior of the membrane as would be expected. Yet, experiments tell us that such cell-penetrating peptides cross the membrane quite rapidly. So what is happening here?
We found that the cell-penetrating peptide can accustom itself to the hydrophobic environment inside the membrane by shedding some of its positive charge. It does this by shuttling some of it protons through a proton wire—essentially a linear chain of water molecules—back to the membrane’s skin or to some negatively charged clusters in close proximity to the skin region. The protons move from the high-potential interior region through the water chain to the low-potential skin region, just like a metal wire connects charge across a potential difference. This effectively leaves behind a neutral peptide that can diffuse more rapidly through the membrane, at a fraction of the energy cost that would otherwise be needed for a cationic peptide. This suggests a chameleon-like behavior through which the peptide camouflages itself in response to changes in hydrophobic-hydrophilic conditions: it loses and then reacquires protons as needed to minimize its free energy within the environment.
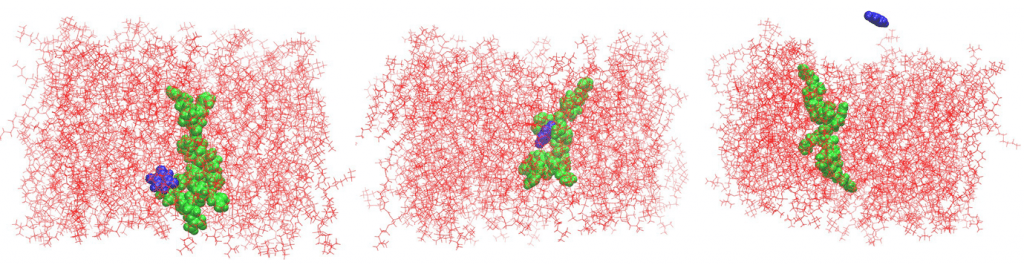
Another revealing outcome from the study is our finding that bridge structures, where a peptide straddles two membrane leaflets, appear to be stable with well-defined anchoring points on both sides of the membrane. This creates migration pathways for small molecules to move across the membrane effortlessly. This phenomena was demonstrated using an anticancer drug 5-fluorouracil (5-FU). The peptide-mediated membrane translocation of the drug molecule (without endocytosis or pore formation) was captured by real-time molecular dynamics. While this won’t make your jaw drop if you are not in the game, it is a staggering observation that has never been reported before by direct molecular simulations.
These new mechanistic insights on how peptides penetrate a membrane are profound, as they offer a new fundamental understanding of how peptides can chemically adapt in two contrasting environments, with far reaching applications in chemical and biological sciences.
Written by:
Sean Smith is Director of the National Computational Infrastructure (NCI Australia) at the Australian National University. He is also Professor of Computational Nanomaterials Science and Technology and heads the Integrated Materials Design Laboratory in the Department of Applied Mathematics, Research School of Physics, ANU.
Hassan Tahini is a principal scientist at the Integrated Materials Design Laboratory in the Department of Applied Mathematics, Research School of Physics, ANU. His research interests lie in the area of computational materials design, with a focus on catalysis and solid-state chemistry.
Reference: Sergio de Luca, et al. ‘Cell Membrane Penetration without Pore Formation: Chameleonic Properties of Dendrimers in Response to Hydrophobic and Hydrophilic Environments‘ Advanced Theory and Simulations (2020). DOI: 10.1002/adts.201900152