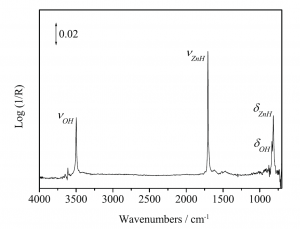
Figure 1. Diffuse reflectance infrared Fourier transform spectrum (DRIFT) of H2 adsorbed on nanocrystalline ZnO at 323K.5
More than half a century ago Eischens, Pliskin and Low discovered that H2 dissociates on the würtzite polymorph of crystalline ZnO.1 The observation of diagnostic infrared active ZnH and OH stretching and bending modes, implied that the H2 splits heterolytically into a surface zinc hydride and a hydroxyl group on a geometrically and electronically anisotropic Zn•••O surface site, to form a proximal hydride and a proton, denoted ZnH•••OH, Figure 1.
At that stage, before the rise of nanochemistry,2 Eischens, Pliskin and Low could not imagine that their pioneering work would inspire the design and construction of surface frustrated Lewis pairs (SFLPs) in a range of solid state materials for a myriad of surface chemistry and catalytic reactions.3,4
Fifty years hence, a great deal more is known about the chemical and physical surface and bulk properties of the würtzite form of ZnO, in single crystal and nanocrystal forms that are deemed responsible for the heterolytic dissociation of H2 on ZnO.5
With the accrued structure and properties knowledge that is available today for the thermodynamically stable hexagonal polymorphic würtzite crystalline form of ZnO, obtained using a battery of diffraction, microscopy, spectroscopy, adsorption and thermal analytical techniques in combination with density functional theory, it is now understood that the energetics and dynamics of the H2 dissociation reaction on the surface of ZnO are crystal face specific and depend sensitively on the type and population of surface defects, in particular the chemical and physical properties of oxygen vacancies.5
In addition, because ZnO alone or in combination with a co-catalyst such as Cu, is an active ingredient for enabling the (reverse) water gas shift reaction and formic acid and methanol synthesis, it has been important to understand how ZnO interacts with the small molecules, CO, CO2, H2, H2O, CH2O, HCO2H and CH3OH, known to participate as reactants, intermediates and products in these catalytic transformations.5
Furthermore, obtaining an in-depth appreciation of the reactivity of different exposed crystal facets and the role of oxygen vacancies in ZnO has proven invaluable in the design, implementation and optimisation of not only ZnO but also other metal oxides and an expanding variety of materials, as high efficiency heterogeneous catalysts for the above reactions.
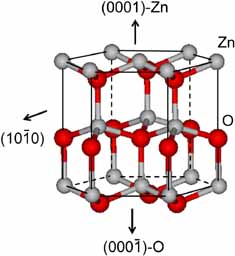
Figure 2. Crystal structure of the würtzite hexagonal polymorphic form of ZnO showing the dominant crystal faces.6
To understand the face specificity of the surface chemistry of ZnO, it is important to examine the anisotropic crystal structure of the stable würtzite polymorphic form, and to look at the different surface sites that exist on the dominant non-polar (101̅0) and polar zinc (0001) and oxygen (0001̅) terminated crystal faces, illustrated in Figure 2.6
The non-polar surface contains equal numbers of close-packed Zn(II) and O(-II) regularly arranged in rows of Zn-O dimers. This surface is electrically neutral and devoid of a surface dipole, to be contrasted with the polar Zn(II) and O(-II) surfaces, which present positive and negative surface charges respectively, with associated surface dipoles. Because the non-polar surface is more stable than the polar ones, different chemical reactivity is anticipated and observed in practice with the above mentioned small molecules. This insight emphasizes the importance of synthetic control of crystal morphology in the design of nanostructured würtzite forms of ZnO, with optimum catalytic activity and the extension of these ideas to other metal oxides.5
An equally important consideration is the formation of oxygen vacancies obtained by removing an oxygen atom from the crystal lattice of stoichiometric ZnO to form non-stoichiometric ZnOx and the resulting effect these vacancies have on the stability and reactivity of the non-polar and polar surfaces of ZnO.5
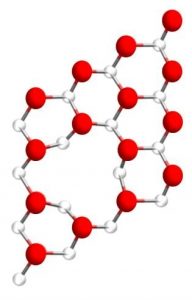
Figure 3. Structure of an oxygen vacancy in a würtzite Zn (0001) surface, red O, white Zn, adapted from Ref. 5.
These vacancies can be created through loss of lattice oxygen by either vacuum thermal treatment or removal as water by thermal reaction with hydrogen. Depending on the number of electrons removed with the lattice oxygen the vacancies can be in a neutral, singly ionized or doubly ionised state, energetically located in the mid-bandgap and induce either semiconducting or metallic character in the non-stoichiometric oxygen deficient ZnOx.5
The structure of an oxygen vacancy formed in a Zn (0001) surface, is illustrated in Figure 3. Compared to a vacancy-free ZnO surface, the Zn(II) sites in ZnOx can be seen to have a higher degree of coordinate unsaturation making it a stronger Lewis acid. Its proximity to Lewis base O(-II) sites provides it with the structure, property and reactivity attributes demanded of what is considered to represent a surface frustrated Lewis pair (SFLP).3,4 Today the SFLP is considered the heterogeneous analogue of the homogeneous frustrated Lewis pair (FLP), the latter also considered to be a story of articulation and exploitation in synthetic molecular chemistry.9-11
To amplify on the history of the FLP before it assumed this descriptor, Halpern in a 1959 paper foresaw the catalytic action for the heterolytic dissociation of H2 on proximal LA-LB pairs, exemplified by Cu(I) or Ag(I) complexes in solution, in particular when the Lewis acidic metal ion is surrounded by Lewis basic ligands or solvent molecules.9
He raised awareness that catalytic activity seemed to require a metal complex with the Lewis acidic metal site in the right valence state and correct surrounding environment. He anticipated the need for “bi-functionality”, in which electron-accepting and proton-accepting sites are simultaneously present – what prescience for today’s FLP. In addition, he realized that some aspects of heterolytic dissociation of H2 on heterogeneous catalysts could also be interpreted with a similar LA-LB model for homogeneous catalytic H2 activation.
In this context, Dayton even earlier in 1953 had solution phase kinetic isotope based mechanistic evidence that forecast the current thinking about FLPs, an insight obtained from the observation of H-D exchange enabled by exposure of D2 to strong alkali metal bases. This exchange chemistry was exemplified by the interaction of D2 with KOH in water and KNH2 in liquid ammonia, both of which have strong Lewis acid K+ sites located in the proximity of strong Lewis base OH– and NH2– sites.10,11
While the origin and the language of the SFLP in materials and the FLP in molecules is different,3,4,8 their chemistry clearly has demonstrated inherent similarities, such as the ability to heterolytically dissociate H2 into a proton and a hydride, respectively bound to the Lewis base LB and Lewis acid LA sites, namely LBH+•••LAH–. This reaction is proving to be a key step in the homogeneous and heterogeneous reduction of gaseous CO2 to energy carriers that include CO, CH3OH, HCO2H, (CH3)2O and CH4.3,4,10
Activation of H2 by the acceptor-donor interactions of the surface Lewis acid Lewis base proximal pair with the sigma bonding and sigma anti-bonding orbitals of H2 serve to weaken and thereby facilitate the heterolytic dissociation of the H-H bond to form ZnH–•••OH+. The hydride and proton can be transferred to the electrophilic carbon and nucleophilic oxygen of gaseous (Eley-Rideal) or adsorbed (Langmuir-Hinshelwood) CO2 to form reduced products, such as methanol or formic acid.3,4
Clearly, the FLP is a new direction in synthetic molecular chemistry and homogeneous catalysis that has a long and recently recognised foundational history in the work of Halpern and Dayton.9-11
Similarly, the SFLP has a long and recently recognized history in surface chemistry and heterogeneous catalysis, which can be traced to pioneering work by Eischens, Pliskin and Low concerning the heterolytic dissociation of H2 on crystalline ZnO,1 the mechanism of which, in the language of today, involves the activation of H2 by a Zn•••O SFLP.3,4
Appreciation of this early pioneering work inspires the design and construction of SFLPs in a range of materials for a myriad of surface chemistry and catalytic reactions based on metal oxides, boron and phosphorous doped silicon nanoparticles, carbon nanotubes, as well as the anchoring of molecular FLPs to the surface of silica nanoparticles, zeolites and metal organic frameworks.
With the wisdom of hindsight, the revelations presented above can be considered an instance of anticipation of FLPs and SFLPs and envisioning their consequences in different classes of molecules and materials. Today with the wisdom of foresight, the attributes of FLPs and SFLPs can now be exploited to advantage in an expanding range of chemical problems involving homogeneous, heterogeneous and enzymatic hydrogenations.
In closing, it is worth mentioning an insightful comment from a colleague who read this opinion editorial, who remarked that this story shows how important it is to revisit or rediscover old work, which because of subsequent developments means that the old discovery or ideas can be used as a springboard for further discoveries or developments. It also begs the question whether enough time is spent on rediscovery or revisiting. Also does the amount of work and papers produced over the last 50 years make it more difficult now to ensure that old work is rediscovered to the benefit of current work? Are there modern tools being developed to assist researchers in rediscovery? Has anyone tried to harness AI to assist in this regard? This whole subject probably could be the subject of a separate article.
References
- P. Eischens, W.A. Pliskin, M.J.D. Low, J. Catal., 1962, 1, 180.
- A. Ozin, Adv. Mater., 1992, 4, 612.
- K. Ghuman, L.B. Hoch, T.E. Wood, C. Mims, C.V. Singh, G.A. Ozin, ACS Catalysis, 2016, 6, 5764 and references cited therein.
- K. Ritter, C&EN, 2016, 94, 26.
- Strunk, K. Kahler, X. Xia, M. Muhler, Surface Science, 2009, 603, 1776 and references cited therein.
- Noei, L. Jin, H. Qiu M. Xu, Y. Gao, J. Zhao, M. Kauer, C. Woll, M. Muhler, Y. Wang, Phys. Status Solidi B, 2013, 250, 1204.
- C. Welch, R.R. San Juan, J.D. Masuda, D.W. Stephan, Science, 2006, 314, 1124.
- W. Stephan, Science, 2016, 354, 1248 and references cited therein.
- Halpern, J. Phys. Chem., 1959, 63, 398.
- K. Wilmarth, J. Dayton, J.M. Fluorny, J. Amer. Chem. Soc., 1953, 76, 4549.
- K. Wilmarth, J. Dayton, J. Amer. Chem. Soc., 1953, 76, 4553.















